CARACTERÍSTICAS CLÍNICAS,
DIAGNÓSTICO Y PRONÓSTICO: (por María Gómez Viciana).
CARACTERÍSTICAS CLÍNICAS:
1. CARACTERÍSTICAS BIOMÉDICAS.
En la etapa de recién nacido, o neonato, y
durante los primeros meses de vida, los bebés presentan el llanto
característico similar al maullido de un gato que cambia de tonalidad a medida
que el niño crece. Además, presentan de un dimorfismo cráneo-facial consistente
en microcefalia, cara redondeada en luna llena, ojos separados, pliegues
epicatales y un puente nasal ancho.
Es común que el niño presente un peso bajo al nacer, inferior a 2,5 Kg., que
es, por lo general, inferior a la media en un 90% de los casos.
Las alteraciones en la talla suelen ser menos marcadas, aunque a lo largo de
las etapas del desarrollo tanto el peso, como la talla y el perímetro craneal,
permanecen inferiores a la media.
Otro aspecto importante en estos niños es el retraso significativo en el
desarrollo psicomotor, apareciendo, en todos los casos, discapacidad
intelectual.
Las malformaciones se ven más detalladamente
en una entrada especialmente centrada en ellas, aquí sólo las estoy enumerando:
|
|
|
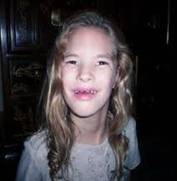
En
esta foto se pueden observar las características predominantes en personas
que padecen este Síndrome: microcefalia, ojos separados, puente nasal ancho,
etc.
|
Características
faciales de un niño con síndrome del maullido del gato a diferentes edades: 8
meses (A), 2 años (B), 4 años (C) y 9 años (D).
|
1.1Malformaciones cráneo- faciales.
1.2. Alteraciones de las extremidades.
1.3. Alteraciones músculo-esqueléticas.
2. CARACTERÍSTICAS PSICOLÓGICAS.
2.1 Características psicomotoras.
En la población afectada por este síndrome,
los hitos evolutivos del desarrollo psicomotor van a ser adquiridos con
posterioridad a lo habitual. A pesar de ello, logros de alta significación
adaptativa como la bipedestación, o el control de esfínteres, son logrados
normalmente sin una gran dificultad.
Por otra parte, en el periodo neonatal y en la primera infancia, presentan una
hipotonía generalizada que, en edades posteriores, puede ser reemplazada por
una hipertonía con reflejos vivos y marcha espástica, mostrando una
coordinación de la motricidad fina y gruesa muy deficitaria.
2.2 .Características cognitivas.
A través de las pruebas psicométricas suele
encontrarse, en estas personas, una discapacidad intelectual que puede llegar a
ser altamente significativa. A pesar de ello, esto no se debe nunca interpretar
como un proceso regresivo, ya que las adquisiciones educativas que se ejercitan
se mantienen y son la base de las adquisiciones posteriores de mayor grado, en
complejidad y funcionalidad.
En esta área cognitiva, también es característica su limitada capacidad de
atención, que deberá ser considerada a la hora de diseñar espacios de
aprendizaje.
Por otro lado, es muy importante que tomemos conciencia de la necesidad que
estos niños presentan de una supervisión permanente, lo cual no debe apartarnos
del objetivo de ayudarle a lograr un grado significativamente bueno de
independencia, con relación a las diversas habilidades de autonomía personal
necesarias.
2.3 . Lenguaje y habla.
A pesar de que estos niños presentan un retraso comunicativo importante, existe
una gran variabilidad respecto a la adquisición del lenguaje y la conducta
verbal.
Por lo general, suelen ser capaces de construir frases sencillas con las que
poder expresar sus necesidades, siendo su capacidad de comprensión verbal
superior a la expresiva.
Por otra parte, el característico llanto de tono agudo y monocorde de estos
niños no posee valor comunicativo significativo.
2.4 .Características conductuales.
Con frecuencia, algunas de las dificultades
que manifiestan en la interacción con su entorno, o en su aprendizaje, son de
carácter instrumental. En este mismo sentido pueden ser frecuentes los
movimientos estereotipados e incluso los comportamientos autodestructivos.
La comunicación fluida entre las familias y los servicios que atienden al niño
van a ayudar a comprender y reconducir estos casos. Este análisis compartido
nos permitirá diferenciar, con seguridad, cuando una conducta es dependiente,
desadaptada o por debajo de las posibilidades de autonomía del niño y cuando
tiene su origen en dificultades para su adquisición, puesta en marcha, o en un
patrón de interacción sustentado en un refuerzo del entorno de alternativas
conductuales inconvenientes. Esta última circunstancia, en muchas ocasiones,
tiene lugar de modo completamente involuntario.
Por último, es preciso decir que no son raras las dificultades para conciliar
el sueño vinculadas a las alteraciones del fondo tónico.
2.5 . Personalidad.
Las dificultades, en el ámbito de la
personalidad están vinculadas al nivel de desarrollo cognitivo y a los
problemas fisiológicos del niño. No obstante, es frecuente encontrar cierto
estado de intranquilidad y nerviosismo generalizado, así como dificultades de
autocontrol emocional.
Su actitud emocional suele ser lábil, viéndose significativamente afectada por
el comportamiento del entorno hacia él. En este sentido, su interés por la
interacción con otros niños es limitado, aunque demuestre gran dependencia de
los adultos, por lo que deben evitarse las tendencias del niño al aislamiento y
estimular su integración y participación.
DIAGNÓSTICO:
El diagnóstico inicial, o de sospecha, es
básicamente clínico ante la presencia de un niño con el llanto característico,
retraso en el crecimiento y las anomalías cráneo-faciales descritas.
La confirmación del diagnóstico se realiza mediante el estudio del cariotipo,
en el que se objetiva la pérdida de un fragmento del cromosoma 5. También,
mediante el uso de técnicas de mapeo cromosómico muy finas, pueden
identificarse las alteraciones específicas en las regiones 5p15.2-5p15.3,
siendo diagnóstico definitivo derivado de dicho estudio cromosómico.
Un tipo de prueba genética más detallada, como el FISH, puede revelar la falta
de una pequeña porción de este cromosoma 5, no detectable mediante el cariotipo
convencional.
El diagnóstico prenatal del síndrome, mediante ecografía, es difícil aunque
puede sospecharse cuando se presentan anomalías graves y un marcado retraso del
crecimiento intrauterino. En este caso podría indicarse una amniocentesis, en
la que se obtendrían células fetales para la realización de un cariotipo en el
que se podría detectar la anomalía cromosómica que confirmaría el diagnóstico.
A día de hoy, la investigación en técnicas de análisis molecular está
permitiendo definir muy precisamente los límites de un cierto número de
deleciones en el brazo corto del cromosoma 5 y comparar los signos clínicos
observados en los pacientes. Esto nos permite explicar la disociación entre los
diferentes elementos del síndrome, para el cual la zona exacta ha podido ser
individualizada.
PRONÓSTICO:
Pronóstico médico.
La esperanza de vida de estos individuos está
disminuida, aunque la mayoría alcanzan la edad adulta (alrededor de los 50
años). Este aspecto depende de la gravedad de las malformaciones asociadas
(cardiopatía…).
La norma es el retraso mental, aunque podemos decir que la mitad de los niños
adquieren las habilidades verbales suficientes para comunicarse en los términos
antes descritos. La estimulación precoz de la comunicación y del área motora ha
mejorado el pronóstico de forma significativa.
Hace años era frecuente que los niños con este síndrome fuesen internados en
instituciones junto a individuos con retraso mental severo. Desde principios de
los años 80 se objetivó que estos niños, integrados en sus familias y con el
beneficio de las técnicas de atención temprana, son capaces de realizar
importantes progresos superando incluso las expectativas de los doctores que
descubrieron por primera vez el síndrome.
Las características físicas de los individuos con este síndrome se vuelven
menos aparentes con el tiempo. El epicantus se atenúa, la cara se alarga, se
hace asimétrica y pierde su redondez. Los huesos faciales comienzan a
modificarse en cuanto a su crecimiento relativo y no se hacen tan evidentes el
hipertelorismo y la micrognatia. El llanto se hace más grave y desaparece el
tono característico al alcanzar el año de edad. La hipotonía tan notable en el
lactante desaparece y los
reflejos se hacen vivos. En la marcha, los pies arrastran por el suelo. El
cabello se hace prematuramente gris. Suelen presentar infecciones de
repetición, otitis medias y dificultades para alimentarse.
A pesar de todos estos avances es todavía difícil ofrecer un pronóstico
individual preciso en las deleciones del brazo corto del cromosoma 5 porque
sujetos portadores de deleciones aparentemente iguales pueden tener fenotipos o
características distintas.
Pronóstico
psicopedagógico.
El pronóstico en estos casos debe ser
realista. La importante discapacidad intelectual establece un marco de
desarrollo que sólo le va a permitir alcanzar una independencia personal
limitada. Sin embargo, nadie puede determinar de antemano cuáles van a ser los
logros de desarrollo que puede alcanzar, por lo que deben tomarse todas
aquellas iniciativas que puedan maximizar su potencial de desarrollo y
posibilitarle la oportunidad de tener una vida plena y lo más normalizada
posible. Por eso, debemos garantizarle los recursos que le posibiliten el mayor
grado de inclusión social y autodeterminación personal.
Los equipos de atención temprana, orientarán a las familias sobre los recursos
y actuaciones de cara a una intervención psicoeducativa a través de programas
de atención temprana que, contra más precoz sea, más eficazmente permitirá
estimular el desarrollo del niño y por lo tanto establecer un pronóstico más
favorable. Dichos programas deben incluir aspectos psicomotrices, sensoriales,
cognitivos, comunicativos, socio-afectivos, así como diseños de modificación de
conducta cuando proceda.
La familia debe obtener el apoyo necesario, no sólo para comprender y aceptar
la realidad de la persona afectada, sino para participar activamente dentro del
programa de atención temprana. Su papel no debe limitarse a cubrir las
necesidades del niño en el ámbito de lo afectivo y asistencial, sino que debe
estar totalmente involucrada con la intervención psicoeducativa diseñada para
favorecer el desarrollo integral de su hijo.
La colaboración de educadores, técnicos y familia es totalmente imprescindible,
pero hay que afirmar que el protagonismo de la familia es singularmente
decisivo durante los primeros años de vida, donde deberán seguir las
instrucciones de estimulación que les marquen los especialistas conforme a su
evaluación de las necesidades y posibilidades concretas cada caso particular.
De cara a esta colaboración, un principio fundamental que ha de tenerse siempre
presente es que las actividades que persigan favorecer el desarrollo del niño
nunca deben de llevarse a cabo con una actitud mecánica, forzada o en
situaciones de cansancio, baja receptividad o desinterés del niño. Por el
contrario, debe cuidarse los aspectos lúdicos y afectivos que van a dar
calidad, significación y potencialidad a la intervención.
Es fundamental que el niño esté motivado y disfrute de las actividades para lo
cual debe evaluarse cuidadosamente la duración de las sesiones más adecuada, el
grado de dificultad que representan para él, nuestras reacciones frente a las
dificultades que pueda manifestar y cómo establecer los pasos sucesivos para
superarlas.
Dentro de las actuaciones de atención temprana orientadas hacia el desarrollo
psicomotor debe considerarse la importancia del masaje, ya que favorece el
desarrollo general del sistema nervioso, la conciencia sensorial y la
corrección del tono muscular característico de este síndrome, al tiempo que
fortalece la comunicación afectiva con el niño, aspecto básico para su
desarrollo.
Dada la tardía aparición de la bipedestación, deben ser estimuladas las formas
previas de desplazamiento, no solamente por la relevancia que tienen de cara a
la coordinación dinámica general del niño, sino por lo que le aproxima a su
entorno y favorece, a través del contacto, exploración y experimentación con
los objetos y personas que le circundan, su desarrollo tanto en el ámbito de lo
cognitivo, como en lo afectivo y social.
En lo relativo al desarrollo cognitivo, éste se va a ver facilitado por
contextos de aprendizaje que le ofrezcan una estimulación multisensorial. No
obstante, un elemento previo y fundamental del programa en este ámbito van a
ser las actividades destinadas a desarrollar las capacidades atencionales del
niño.
Lógicamente, en este sentido, el objetivo inicial debe de ser el
establecimiento y mantenimiento del contacto visual, primero hacia las personas
que cuidan de él y luego sobre modificaciones contextuales que introducimos en
su entorno, así como sobre los objetos con los que está llevando a cabo una
actividad. Facilita este propósito el uso de materiales atractivos y novedosos,
junto a la eliminación de aquellos otros estímulos que distraigan su atención.
En cuanto a su capacidad comunicativa, sabemos que, en general, sus primeras
palabras aparecen entre los 3 y los 6 años y que, aunque algunos podrán
construir oraciones completas, en otros casos sólo serán capaces de utilizar
unas pocas palabras aisladas, puede estar indicado el aprendizaje de métodos
alternativos de comunicación.
En cualquier caso, su capacidad comunicativa debe ser objeto de constante
estimulación, favoreciendo la complementación de las expresiones verbales y
gestuales, aprovechando para ello todas las circunstancias cotidianas de
interacción para acompañarlas y enriquecerlas con expresiones verbales e
impulsar la adquisición y uso por parte del niño de los elementos lingüísticos
en sus comunicaciones, aportándole ejemplos adaptados, ocasiones de utilización
y estímulo ante sus ejecuciones.
No debemos olvidar a este respecto que la estimulación de su capacidad
comunicativa debe apoyarse siempre en los valores funcionales de la misma, si la
comunicación no facilita al niño la expresión de sus necesidades y la
satisfacción de sus deseos, no encontrará justificación para esforzarse en
ella.
En general, a la hora de diseñar el programa debe considerarse especialmente la
funcionalidad de los aprendizajes planteados de cara a la mejor inclusión en su
entorno. Con relación a ello, adquiere un papel prioritario la adquisición de
hábitos y actitudes que faciliten su autonomía personal, estableciendo
contextos de aprendizaje progresivos para este fin.
Además, es conveniente tener en cuenta la madurez necesaria para el inicio de
cada actividad y, por lo tanto, si el niño concreto precisa aprendizajes
previos antes de abordarla. Es importante adecuar el ritmo de aprendizaje a
cada niño y la correcta consolidación de cada paso a la hora plantearse un
nuevo objetivo.
A medio plazo podemos establecer los siguientes elementos como fundamentales de
cara a maximizar las posibilidades de autodeterminación, autonomía personal e
inclusión social del individuo:
- Independencia en relación con aspectos básicos del aseo e higiene personal,
control de esfínteres, ciclos de vigilia-sueño apropiados y estables, pautas de
alimentación.
- Autonomía o colaboración activa respecto al manejo del vestuario y limpieza
de su entorno vital.
- Hábitos en la mesa y en otras situaciones afectadas por los usos y costumbres
de su entorno social.
- Trato a las personas en todo tipo de situaciones sociales.
- Cuando sea posible, formación pre-laboral.
Debemos prestar especial cuidado en favorecer
que los aprendizajes que se han llevado a cabo no se extingan por falta de
ocasiones para ponerlos en práctica, así como posibilitar que sean
generalizados a otros contextos reales donde puedan servir para facilitar la
integración social del individuo.
Las técnicas de modificación de conducta son eficaces y pueden servirnos de
ayuda, tanto para la instauración de estas habilidades, como para la
intervención frente a comportamientos autodestructivos o inadaptados. Sin
embargo, es imprescindible en estos casos que todas las personas que rodean al
niño acuerden una misma respuesta frente a cada circunstancia. Algunas de las
dificultades respecto a la alimentación en relación con la succión, babeo,
deglución o masticación, debidas a la inicial hipotonía pueden abordarse
también por esta vía.
Un objetivo prioritario con estos niños dentro del ámbito psicosocial es
potenciar su autocontrol emocional. Para ello, debe fortalecerse su autoestima
a través de nuestra valoración y afecto, así como el ejercicio de la
responsabilidad a la medida apropiada de sus posibilidades.
Su integración y participación social están muy vinculadas a la adquisición de
hábitos y habilidades de interacción, que a su vez van a ser sus recursos
fundamentales para el autocontrol emocional en los contextos sociales. Estas
habilidades permitirán su normal participación en las actividades que se estén
llevando a cabo en su entorno, posibilitarán su aceptación por otras personas y
evitarán su aislamiento.
El juego es una estrategia esencial a través de la que estimular el desarrollo
psicosocial de estos niños. A través de él, se potencian los vínculos afectivos
y sociales, al tiempo que se posibilitan las experiencias sensorio-motoras que
sustentaran su desarrollo cognitivo. Además, en el contexto del juego, el
aprendizaje por observación se ve potenciado de forma significativa, siendo
básico dicho tipo de aprendizaje para el desarrollo social del niño.
Inicialmente el juego no va a involucrar objetos. Deberá tratarse de una
interacción gratificante con el niño, donde el contacto físico y los gestos de
sus cuidadores son decisivos. Todas las actividades cotidianas, como
alimentarle o bañarle deben tener un componente lúdico y de comunicación
afectiva. Cuando su desarrollo se lo permita, han de entrar en escena los
objetos que le van a permitir explorar las características de su entorno físico
y avanzar en su desarrollo cognitivo.
El momento de la escolarización también va a ser decisivo a este respecto, ya
que le va a permitir participar de un entorno social. Su momento de inicio ha
de ser valorado considerando las orientaciones de los especialistas que hacen
el seguimiento del caso, escogiéndose el centro donde se va a llevar a cabo,
considerando los servicios y recursos de que dispone. Su posterior
escolarización en las etapas obligatorias debe contar con una evaluación
psicopedagógica y un dictamen de escolarización realizado por los servicios
competentes de la administración autonómica.
Con carácter general dicha escolarización ha de ser lo menos restrictiva
posible, considerando siempre las características del caso particular y las
necesidades que de ellas se derivan. Cuando los centros específicos sean la
opción más aconsejable, debe buscarse entornos complementarios donde el niño
pueda integrarse en actividades de ocio y tiempo libre con otros niños en un
contexto de integración.
Finalizando la escolaridad obligatoria, debe prepararse para una futura
inserción sociolaboral a través de un centro protegido de empleo.
Pautas para el aprendizaje:
- Las actividades tendrán, como se ha dicho
anteriormente, un carácter lúdico, con materiales atractivos.
- Ambiente tranquilo y relajado.
- Confección de libretas con imágenes (preferentemente reales) para la
identificación de acciones, situaciones, personas…
- Buscar el momento en el que el niño esté más receptivo.
- La presentación de actividades se hará gradualmente: de sencillas a
complicadas.
- Basarnos en la motivación del niño, sin forzarle.
- Dejarle probar y experimentar.
- Ofrecerle la posibilidad de jugar libremente.
- Respetar los tiempos de aprendizaje.
- Tener en cuenta los diferentes ritmos de adquisición de los aprendizajes.
Bibliografía:
http://www.asimaga.org/estudio-psicopedagogico-del-sindrome-del-maullido-del-gato/
http://es.wikipedia.org/wiki/S%C3%ADndrome_del_maullido_de_gato
http://www.nlm.nih.gov/medlineplus/spanish/ency/article/001593.htm